|
|
|
UN TEST CHE NON SBAGLIA Le vicende doping legate alla diffusione nello sport del C.e.r.a., l'epo di terza generazione, hanno messo in rilievo voci e polemiche sul test recentemente adottato dal laboratorio di Chatenay Malabry; uno dei piů avanzati nel mondo nella lotta all'epo, le cui tecniche e procedure sono universalmente riconosciute. Dal professor Dario D'Ottavio, eminente biochimico clinico, nonché collaboratore di numerose Procure d'Italia nelle indagini doping, riceviamo questo prezioso contributo teso a chiarire in concreto come funziona questo metodo che qualcuno vuole contestare. Si tratta di un articolo molto tecnico, ma anche molto chiaro da cui emergono sostanzialmente tre fatti concreti: 1) il risultato del test, per
i riscontri scientifici che offre, č altamente specifico. Cioč non ci sono
dubbi sul tipo di molecola individuata.
|

|
|
di Dario D'Ottavio PROTEINE, ISOFORME E PUNTO ISOELETTRICO Le proteine sono
la componente principale sia qualitativa che quantitativa del
sangue. Esse sono costituite da una sequenza di amminoacidi
legati l’un altro. Altresě questa sequenza puň essere coniugata con
ulteriori gruppi o metalli. Inoltre č da rilevare che la stessa
proteina, a livello plasmatico, puň essere presente in differenti
“isoforme” che sono caratterizzate dalle stesse proprietŕ
biochimiche, ma con leggere differenze chimico fisiche dovute, per
lo piů, a modificazioni post traduzionali. Le proteine del sangue
differiscono per dimensioni, forma e carica elettrica; sono anfotere
(come i peptidi o gli aminoacidi) e possono avere a seconda del pH
del mezzo che fa da supporto, o carica negativa o carica positiva o
carica nulla. Per ciascuna proteina, esiste quindi un valore di
pH riferendosi al quale la risultante delle cariche positive e
negative č nulla, ovvero la proteina č elettroforeticamente
immobile, ciň che viene definito comunemente “punto
isoelettrico”. LE PROTEINE PLASMATICHE (SANGUE) Tiselius, nel lontano 1937, per primo, mise a punto un metodo di migrazione e separazione delle proteine in un campo elettrico, utilizzando un supporto liquido; successivamente carta, acetato di cellulosa, gel d’agar, agarosio vennero utilizzati come mezzi stabilizzanti per “intrappolare” le proteine sostituendo cosě il supporto liquido. Le proteine plasmatiche, sottoposte ad elettroforesi, danno origine ad una serie di 6 bande (fibrinogeno compreso) denominate Albumine, α1, α2, β1,β2 e gamma globulina. Se pur di notevole interesse diagnostico (oggi con l’elettroforesi capillare si riescono ad ottenere decine di bande) il limite di questa tecnica č quello di raggruppare in ciascuna banda molteplici tipi di proteine caratterizzanti patologie diverse. IDENTIFICAZIONE DELLE PROTEINE La caratterizzazione, cioč l'identificazione della singola proteina avviene allorché si accoppia all’elettroforesi classica la reazione antigene – anticorpo che si identifica nell’immunoelettroforesi. Esemplificando, si fanno migrare le proteine in un campo elettrico e successivamente si mettono a contatto per diffusione con un anticorpo specifico per la proteina di interesse; qualora la proteina fosse presente, nella zona corrispondente alla posizione di migrazione si noterŕ un precipitato dovuto alla reazione antigene anticorpo. Il limite di questa tecnica consiste nel fatto che affinché avvenga la reazione antigene anticorpo con precipitazione del complesso formatosi debbono essere rispettati precisi rapporti di concentrazione tra le due sostanze e quindi, per rilevare proteine a basse concentrazioni, questa tecnica diventa improponibile. Ulteriori passi avanti sono stati fatti con l’ausilio di anticorpi “marcati” con un atomo radioattivo o un gruppo fluorescente o chemiluminescente oppure coniugati con enzimi particolari capaci di dare origine a reazioni colorimetriche; tutti sistemi che ampliano l’intervallo di concentrazione in cui le singole proteine possano essere differenziate. I MIGLIORAMENTI: TAMPONI ALCALINI E IMMUNOELETTROFOCOUSING Miglioramenti della selettivitŕ delle bande sono stati ottenuti passando dall’uso di tamponi alcalini a pH fisso (in genere compreso tra 8-9) all’uso di anfoliti che consentissero di ottenere un gradiente di pH. In questo caso, oltre alla separazione dovuta al differente peso molecolare delle varie proteine, queste si posizioneranno nelle zone ove il pH č uguale al punto isoelettrico e quindi la velocitŕ di migrazione č praticamente nulla: parliamo in questo caso di elettrofocusing o immunoelettrofocusing se si utilizzano anticorpi specifici. Le tecniche si sono via via sempre piů affinate cercando di ridurre od eliminare le reazioni aspecifice tra anticorpo e proteina (cross – reaction) responsabili di false positivitŕ in quanto, data la molteplicitŕ di proteine presenti nel plasma, una reazione aspecifica (specie con proteine presenti in elevata concentrazione) č pur sempre possibile. TOBWIN E IL "BLOTTING" Introdotta nel 1979 da Towbin, cominciň a diventare routine nei laboratori di ricerca la tecnica di “blotting” tutt’oggi comunemente utilizzata per determinare piccole quantitŕ di proteine presenti in matrici complesse quali il plasma ed il siero umani. La semplice procedura di “blotting” (conosciuta come “blot” o “slot blot”) si basava su una filtrazione sotto vuoto per trasferire le proteine su una membrana a micropori. Mentre questo metodo fornisce informazioni qualitative sull’espressione dei livelli totali delle proteine e puň essere utilizzato per campioni multipli in parallelo, difetta dell’informazione sul loro peso molecolare. Inoltre, la specificitŕ puň essere compromessa dalla degradazione delle proteine qualora fosse necessaria la loro integritŕ. Una applicazione moderna di quanto precedentemente detto č la “Western blotting” che implica la separazione elettroforetica delle proteine, con successivo elettrotrasferimento delle stesse su una idonea membrana. In particolare, le proteine vengono separate attraverso una elettroforesi su gel. Questa separazione puň essere ottenuta o tramite il loro punto isoelettrico o il peso molecolare o la carica elettrica, in genere si usa una combinazione di questi tre fattori. Il piů comune tipo di gel elettroforesi utilizza un gel di poliacrilamide e un tampone ricco di dodecilsolfato sodico (SDS) che mantiene i polipeptidi in uno stato denaturato una volta che sono stati trattati con forti agenti riducenti per rimuovere la struttura secondaria e terziaria (ad es. i gruppi disolfuro (-S-S-) in modo di separare le proteine secondo il loro peso molecolare. In questa situazione le proteine si caricano negativamente e migrano verso il polo posititivo attraverso il reticolo dell’acrilamide allo stato di gel. Le proteine piů piccole migreranno piů rapidamente attraverso i pori del gel e cosě si separeranno a seconda del loro peso molecolare. La concentrazione di acrilamide determina la risoluzione del gel – maggiore č la sua concentrazione e maggiore sarŕ la risoluzione per proteine a basso peso molecolare. Quindi dopo aver depositato le proteine sul gel ed applicato il voltaggio, le proteine si muoveranno nel gel con differenti velocitŕ; questo diverso stato di avanzamento (differente mobilitŕ elettroforetica) č quello che determina la separazione in bande all’interno di ogni zona di deposito. REAZIONE CON ANTICORPO SPECIFICO ED "ELETTROBLOTTING" Affinché le proteine cosě separate possano reagire con l’anticorpo, debbono essere rimosse dal gel e trasferite su una membrana di nitrocellulosa o di difuoropolivinilidene. La membrana viene posta al di sopra del gel e il tutto coperto da carta da filtro. L’insieme viene poi posto in una soluzione tampone che sposta verso l’alto la carta per azione capillare portando con se le proteine trasferendole sulla membrana. Altro metodo per trasferire le proteine č l’elettroblotting che utilizza la corrente elettrica per trasferire le proteine su una membrana in PVDF o nitrocellulosa. Le proteine passano dal gel alla membrana mantenendo le stesse posizioni assunte sul gel. Come risultato di questo processo di “blotting” le proteine sono ora depositate su una superficie sottile ottimale per la loro determinazione. Dato perň che la membrana č stata scelta per la sua capacitŕ di legare le proteine ed entrambi gli anticorpi, successivamente utilizzati, sono proteine, bisogna prevedere un ulteriore fase per prevenire che quest’ultimi vadano ad occupare gli spazi liberi della membrana. Ovvero il “bloking” della membrana per evitare legami aspecifici, che si effettua ponendo questa in una soluzione diluita a contenuto proteico (generalmente albumina sierica bovina o caseina) in modo tale che questi composti vanno ad eliminare la possibilitŕ che gli anticorpi utilizzati possano specificamente legarsi alla membrana. Questo processo, in definitiva, serve ad eliminare i falsi positivi. ANTICORPI PRIMARI E SECONDARI L’evidenziazione della proteine di interesse viene effettuata utilizzando un primo anticorpo “primario” e un secondo anticorpo “secondario”. Il primo anticorpo č specifico per la proteina da determinare e dopo il “bloccaggio” della membrana viene messo a contatto con quest’ultima per i periodo di tempo necessario all’espletamento della reazione di legame. Dopo un opportuno lavaggio per eliminare l’eccesso di anticorpo “primario” (ovviamente non legato alla proteina ricercata e comunque ancora in grado di reagire) , la membrana viene trattata con un anticorpo “secondario” specie-specifico di una porzione dell’anticorpo “primario” precedentemente utilizzato. L’anticorpo secondario č generalmente legato a biotina od a un enzima, in genere fosfatasi alcalina e perossidasi. La biotina o l’enzima , attraverso una serie di reazioni formano un composto che rende “visibili” le bande delle proteine che hanno reagito con l’anticorpo primario. La posizione della bande e la loro intensitŕ sono caratteristiche peculiari di ogni specie proteica. La tecnica descritta consta quindi di due fasi: legame della proteina con l’anticorpo specifico e successivo legame del complesso antigene –anticorpo con un anticorpo marcato; tele processo č definito two-step process. E’ comunque possibile, qualora si utilizzi un anticorpo specifico giŕ “marcato”, poter effettuare la determinazione in un unico step. Tale processo č piů rapido ma non č scevro da possibili interferenze per cui viene utilizzato soltanto per lo “screening” dei campioni positivi. IL DOUBLE BLOTTING Lasne nel 2001 ha sviluppato un metodo “Double blotting” per superare il problema di legami non specifici da parte dell’anticorpo secondario nell’immunoblotting. In pratica, dopo che č stato applicato l’anticorpo primario, la membrana con le proteine “blottate” viene assemblata con una seconda membrana “pulita” e sottoposta ad un secondo “blotting” sotto condizioni acide. Le molecole dell’anticorpo primario sono cosě desorbite dal loro corrispondente antigene e trasferite sulla seconda membrana, mentre l’antigene e le proteine interferenti restano legate alla prima. La seconda membrana puň quindi essere trattata con l’anticorpo secondario senza il rischio di legami aspecifici. Questo tipo di metodica č stato sviluppato per lo studio dell’eritropoietina in urine concentrate poiché sono stati evidenziati forti legami non specifici con gli anticorpi biotinilati in alcuni campioni sottoposti ad analisi. Da quanto sopra si puň dedurre che l’applicazione dell’immunoettrofocusing “double blotting” č altamente specifica per la ricerca della proteina di interesse, presenta comunque il difetto che in presenza di piccole quantitŕ, dato che in ogni passaggio si ha inevitabilmente perdita di materiale, che proteine a bassa concentrazione possano ancor piů diluirsi e quindi non reagire con l’anticorpo secondario dando origine a “falsi negativi”. |
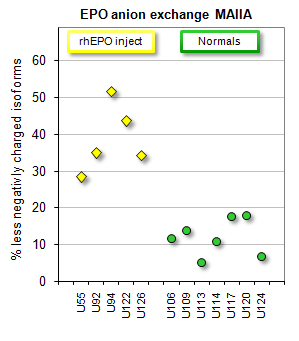 |
Il test della Maiia disgnostic consente di distinguere l'epo ricombinante (esogena) da quella endogena, prodotta dal fisico umano attraverso l'identificazione della carica diversa. Le isoforme dell'epo ricombinante (puntini gialli) sono meno negative di quelle endogene (verdi). |
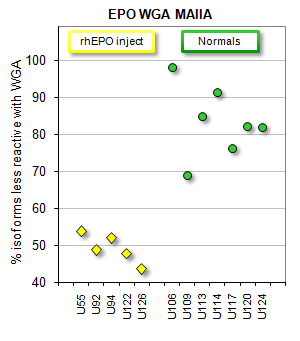
Un test ulteriore (Epo Wga) distingue l'epo ricombinante (esogena) da quella endogena attraverso la loro differente interazione con la lattina. Il legame della forma endogena di epo (puntini verdi) č piů debole di quello della forma esogena (puntini gialli). |
 |
|
Questo test (EPO MAIIA) consente di distinguere le isoforme dell'epo endogena da quelle esogene (assunzione esterna) come l'epoetina alpha, beta, omega, delta, e forme analoghe come la darboepoetina alfa e il CERA. Anche se l'epo appare in basse concentrazioni nell'urina. Il metodo attuale, accreditato dalla Wada, cioč riconosciuto come valido per rintracciare l'epo esogena dall'agenzia mondiale antidoping, combina l'isoelectric focousing (IEF) con il "double immunoblotting". E' stato descritto da F. Lasne e J. de Ceaurriz su "Nature", la piů prestigiosa delle riviste scientifiche, nel 2000 (Nature 405, 635). |
|||
![]()